

Galactosémia clássica
Introdução
Definição
As galactosémias são doenças rara, de transmissão autossómica recessiva, resultantes da alteração do metabolismo da galactose, por ocorrência de um defeito numa das enzimas intervenientes na via de degradação deste monossacarídeo (1).
Epidemiologia
A galactosémia clássica, a forma menos rara das galactosémias, tem uma incidência de 1:23000-44000 recém-nascidos (RN) na Europa. Na Irlanda a incidência é cerca de 1:20000 RN. Na população asiática esta doença é mais rara. Em Portugal, a sua prevalência é desconhecida. No nosso país, o rastreio neonatal (teste de pezinho) não inclui o seu rastreio, contrariamente ao que ocorre em muitos países europeus.
Fisiopatologia
A lactose, o principal hidrato de carbono do leite, é um dissacarídeo formado por glicose e galactose. É hidrolisada em glicose e galactose por ação da lactase da superfície intestinal.
Após a absorção, a galactose é metabolizada em glicose 1-fosfato pela ação de três enzimas consecutivas (Figura 1). O defeito em qualquer uma destas enzimas pode causar galactosémia, ie aumento da concentração de galatose no sangue. O mais frequente é o da GALT (galactose 1-fosfato uridiltransferase), que causa a galactosémia clássica.
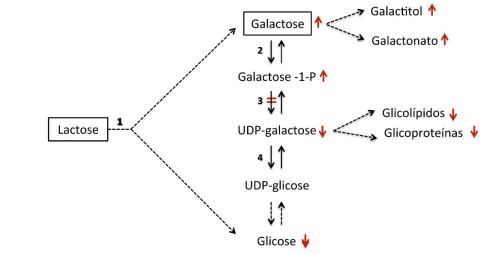
Quando a atividade da GALT está diminuída, os substratos acumulam-se em níveis tóxicos (galactose e galactose-1-fosfato), enquanto os produtos se tornam deficientes (UDP-galactose). São também ativadas vias acessórias de metabolização da galactose, com formação de produtos em doses tóxicas, como o galactitol e galactonato (Figura 1).
Níveis elevados de galactose e galactose-1-fosfato causam lesão hepática e renal. A acumulação de galactitol no cristalino origina cataratas. A UDP-galactose é precursora de oligossacarídeos que compõem as glicoproteínas e os glicolípidos. A sua diminuição leva a uma alteração secundária da glicosilação, contribuindo, nomeadamente, para as complicações que ocorrem a nível do sistema nervoso central e ovárico (1).
A galactosémia clássica é uma doença de hereditariedade autossómica recessiva. Conhecem-se mais de 250 mutações, sendo a mais frequente a p.Q188R, no gene GALT (70%). Esta mutação é a mais comum em caucasianos e na Europa do Norte. Está bem estabelecida uma correlação genótipo-fenótipo, sendo a mutação anteriormente referida associada a um prognóstico neurológico pior (2).
História Clínica
Apresentação clínica
A galactosémia clássica surge após o RN iniciar a alimentação com leite. Manifesta-se geralmente nas primeiras semanas de vida, mas os sintomas podem ser muito precoces, surgindo logo no 3º-4º dia de vida. Os sintomas podem ser atenuados se a alimentação com leite for interrompida no caso de o RN iniciar fluidoterapia endovenosa. Os RN podem apresentar sintomatologia de intolerância alimentar com dificuldade/recusa alimentar, vómitos, má progressão/perda ponderal e/ou não recuperação do peso de nascimento. Por outro lado, o quadro inicial pode ser indistinguível ao de outra hepatopatia, podendo haver progressão rápida para insuficiência hepática ou para cirrose. A lesão renal também é frequente, ocorrendo perda urinária de galactose, glicose, fósforo, aminoácidos (tubulopatia proximal ou síndrome de Fanconi). Há uma suscetibilidade a infeções por bactérias Gram negativas (Escherichia coli). Alguns doentes (10%) apresentam um quadro de sépsis, que pode ser fatal.
Exame objectivo
No exame objetivo podem ser evidentes sinais de desnutrição e hepatopatia, nomeadamente, icterícia, hepatoesplenomegalia, distensão abdominal, ascite, e evidência de alterações da coagulação (hematomas ou hemorragia após uma simples colheita de sangue).
Os RN podem apresentar abaulamento da fontanela anterior (pesudotumor cerebri), letargia e hipotonia.
As cataratas, embora raramente presentes ao nascimento, podem surgir muito precocemente.
Diagnóstico Diferencial
De acordo com o referido anteriormente e tendo em conta a forma de apresentação, deve ser feito o diagnóstico diferencial com sépsis neonatal, bem como com causas de insuficiência hepática neonatal (3).
Exames Complementares
Patologia Clínica
O diagnóstico desta doença passa pelo reconhecimento dos sintomas após o início do aleitamento, associado ao diagnóstico bioquímico (doseamento de metabolitos tóxicos, determinação da atividade enzimática) e genético.
Laboratorialmente há evidência de lesão hepática (hiperbilirrubinémia não conjugada, elevação das transaminases, coagulopatia, elevação de alguns aminoácidos plasmáticos como acfenilalanina, a tirosina e a metionina), de doença tubular renal (aumento da excreção urinária de galactose, glicose, fósforo, aminoácidos), acidose metabólica com hiato aniónico normal (hiperclorémica, por perda renal de bicarbonato), hipofosfatémia, anemia hemolítica. Menos frequentemente, pode surgir hipoglicemia.
A pesquisa de açúcares redutores na urina é um método de rastreio, mas não confirma ou exclui o diagnóstico. Pode apresentar falsos positivos: excreção urinária de outras substâncias redutoras (ex. glicose); situações de falência hepática aguda de outra etiologia, com acumulação de galactose. Os falsos negativos surgem na presença de jejum prolongado (>24-48h) e/ou no caso do RN estar sob fluidoterapia endovenosa ou apresentar recusa alimentar/vómitos incoercíveis.
A suspeita clínica pode ser demonstrada pela elevação de galactose e galactose-1-fosfato no sangue e urina e de galactitol na urina.
O gold standard para o diagnóstico é o estudo da atividade enzimática da GALT nos eritrócitos. É necessário confirmar que o RN tenha sido submetido a transfusão de glóbulos vermelhos, pela interferência na interpretação dos resultados, caso em que se deve adiar o estudo enzimático.
A confirmação do diagnóstico é feita pelo estudo do gene GALT.
Tratamento
O tratamento requer a evicção da galactose da dieta. Logo que surja a suspeita de galactosémia, o leite, materno ou de fórmula deve ser suspenso de imediato e substituído por um leite sem galactose, mesmo que a pesquisa de açúcares redutores seja negativa, e ainda antes serem conhecidos os resultados dos exames complementares de diagnóstico.
Desde que introduzida precocemente, a interrupção do consumo de galactose reverte rapidamente a lesão hepática e renal. O mesmo acontece com as cataratas na maior parte dos casos. Na fase aguda, pode ser necessário terapêutica adicional de suporte da doença hepática, dos desequilíbrios hidroeletrolítcos, da acidose metabólica e de eventual sépsis associada.
Aquando da diversificação alimentar, a criança deverá evitar os alimentos que contenham lactose/ galactose (vísceras - fígado, rim e mioleira, legumes - lentilhas, grão de bico, algumas frutas). Os pais e os doentes devem ser instruídos a verificar os rótulos dos alimentos, bem como a composição dos excipientes dos medicamentos.
Deve ser feita suplementação com vitaminas (D, K) e iões (cálcio) de forma a repor défices provenientes da dieta restritiva a que o doente ficará sujeito.
Na fase de suspeita da doença deve, a par da suspensão alimentar da galactose, ser feito o contacto/ referenciação com um Centro de Referência de Doenças Hereditárias do Metabolismo, de modo a que o diagnóstico etiológico, tratamento e seguimento do doente sejam os mais adequados, dada a raridade da doença, de acordo com a norma 012/2017 da Direção Geral da Saúde (Abordagem Diagnóstica e Critérios de Referenciação de Doenças Hereditárias do Metabolismo em Idade Pediátrica e no Adulto, em: dqs@dgs.min-saude.pt).
Evolução
Independentemente do cumprimento da dieta restrita em galactose podem surgir complicações tardias, nomeadamente, atraso de desenvolvimento psicomotor/défice cognitivo, dificuldades de aprendizagem, alterações da linguagem, alterações neurológicas (tremor, disartria, ataxia, manifestações extra-piramidais), disfunção gonadal, sobretudo no sexo feminino (atraso pubertário, insuficiência ovárica secundária), atraso de crescimento (baixa estatura) e osteopenia.
Uma vez que a insuficiência ovárica é secundária e poderá ser equacionada a reserva de óvulos na puberdade para utilização futura (1). Assim, após a puberdade as doentes devem ser seguidas em Consultas de Ginecologia e Aconselhamento Genético.
O quadro de apresentação inicial pode ser grave e colocar a vida em risco por sépsis, insuficiência hepática/cirrose. Ultrapassada esta fase, o prognóstico dependerá do aparecimento das complicações tardias que não são evitáveis com a restrição da galactose na dieta, pelo que recentemente se tem equacionado a necessidade do seu rigor (4). Pensa-se que esta evolução se relaciona com a exposição pré-natal à galactose e com a produção endógena de galactose a partir da glicose que vai ocorrendo ao longo da vida.
Recomendações
O seguimento destes doentes deve ser feito por equipas multidisciplinares, que incluam médicos e nutricionistas com experiência em Doenças Hereditárias do Metabolismo.
Deve ser feita a monitorização do cumprimento da terapêutica e evolução. Os doseamentos dos níveis plasmáticos de galactose-1-fosfato (ideal
Simultaneamente, deve ser feita a vigilância de eventuais complicações, o que inclui a avaliação oftalmológica anual, avaliação do estadio pubertário e doseamentos hormonais (estradiol, FSH, LH) e a monitorização do desenvolvimento psicomotor/ intelectual e rendimento escolar com apoio de Psicólogo. Tratando-se de uma doença crónica, com importante impacto na qualidade de vida, os aspetos psico-sociais no seguimento dos doentes e famílias não devem ser descurados (5).
Deve ser oferecido aconselhamento genético aos pais. A galactosémia é uma doença genética, autossómica recessiva. Os pais são, em princípio, portadores do defeito enzimático; podem transmiti-lo, mas não sofrem da doença. Numa gravidez subsequente, a probabilidade de recorrência da doença é de 25%. É possível fazer o diagnóstico pré-natal, preferencialmente através do estudo genético em biópsia de vilosidades coriónicas, embora também seja possível fazê-lo através da determinação da atividade da GALT e o doseamento de galactitol (1).
Bibliografia
- Berry GT, Walter J, Fridovich-Keil JL (2016) Disorders of galactose metabolism. In: Saudubray JM, van den Berghe G, Walter JH (eds) Inborn metabolic diseases: diagnosis and treatment. Springer, Heidelberg
- Coelho AI, Ramos R, Gaspar A, Costa C, Oliveira A, Diogo L, Garcia P, Paiva S, Martins E, Teles EL, Rodrigues E, Cardoso MT, Ferreira E, Sequeira S, Leite M, Silva MJ, de Almeida IT, Vicente JB, Rivera I. A frequent splicing mutation and novel missense mutations color the updated mutational spectrum of classic galactosemia in Portugal. J Inherit Metab Dis. 2014 Jan;37(1):43-52.
- Dias Costa F, Moinho R, Ferreira S, Garcia P, Diogo L, Gonçalves I, Pinto C. Acute liver failure related to inherited metabolic diseases in young children. An Pediatr (Barc). 2017 Apr 11
- Frederick AB, Cutler DJ, Fridovich-Keil JL. Rigor of non-dairy galactose restriction in early childhood, measured by retrospective survey, does not associate with severity of five long-term outcomes quantified in 231 children and adults with classic galactosemia. J Inherit Metab Dis. 2017 Nov;40(6):813-821.
-
Hoffmann B, Dragano N, Schweitzer-Krantz S (2012) Living situation, occupation and health-related quality of life in adult patients with classic galactosemia. J Inherit Metab Dis 35:1051–1058.
Deseja sugerir alguma alteração para este tema?
Existe algum tema que queira ver na Pedipedia - Enciclopédia Online?