

Deficiência de piruvato desidrogenase
Introdução
Definição
A deficiência de piruvato desidrogenase (DPDH) é uma doença rara do metabolismo energético da glicose. A PDH é um complexo multienzimático localizado na matriz mitocondrial. Estão descritos 6 genes nucleares associados a esta doença, com hereditariedade ligada ao cromossoma X (na quase totalidade dos casos) ou autossómica recessiva (1).
Epidemiologia
A DPDH é uma doença muito rara, com incidência e prevalência desconhecidas (1). Apesar da raridade, é uma das principais causas de acidose láctica congénita. O seu diagnóstico é habitualmente evocado com base na clínica, predominantemente neurológica, não estando incluída no rastreio neonatal.
Fisiopatologia
O complexo PDH catalisa a descarboxilação oxidativa do piruvato, o principal produto da glicólise, originando acetil-coenzima A (acetil-coA), que é posteriormente oxidada no ciclo de Krebs. A energia derivada da oxidação completa da glicose, via piruvato, é usada na cadeia respiratória mitocondrial para a síntese de ATP, a molécula de troca da energia química. Quando há uma deficiência da PDH, ocorre um bloqueio na oxidação aeróbica do piruvato, com aumento da produção de lactato e alanina (figura 1). Assim, há um défice energético associado a acumulação de piruvato, lactato e alanina no sangue, urina e líquido cefalorraquidiano, agravada pelo consumo de carbohidratos. Estes são desaproveitados do ponto de vista energético, uma vez que a glicólise passa a ser “anaeróbica”, apesar da disponibilidade normal de oxigénio (2).
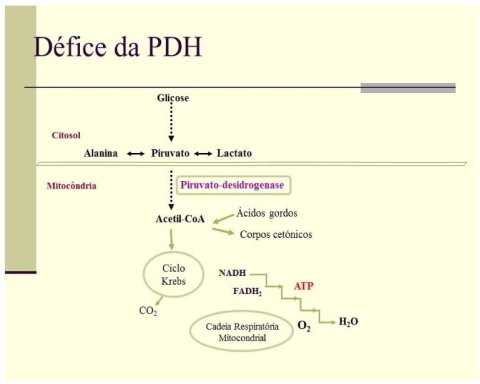
O complexo da PDH é constituído por três subunidades enzimáticas: piruvato desidrogenase (E1) – alfa e beta –, dihidrolipoamida transacetilase (E2) e dihidrolipoamida desidrogenase (E3), e uma proteína de ligação da E3. A sua atividade, estimulada pela PDH fosfatase, necessita de coenzimas: tiamina, ácido lipóico e nicotinamida-adenina-dinucleótido (NAD+).
A maioria dos casos de DPDH é causada por mutações no gene PDHA1, o único localizado no cromossoma X, que codifica a subunidade E1 alfa. Está associado a hereditariedade ligada ao X dominante ou recessiva, conforme o tipo de mutação e o padrão de inativação do X (nos doentes do sexo feminino). (1). Estão descritas cerca de 140 mutações no gene PDHA1 (1, 3).
Mais raramente foram descritas mutações associadas a hereditariedade autossómica recessiva nos genes que codificam para as outras subunidades da PDH e para a PDH fosfatase. O gene LIPT1, interveniente na via de síntese do ácido lipóico, foi igualmente associado a DPDH (4).
História Clínica
Apresentação clínica
A DPDH apresenta-se com quadros predominantemente neurológicos, sem hipoglicemia. A grande heterogeneidade genética explica a elevada variabilidade fenotípica. Esta vai desde formas neonatais com envolvimento neurológico e acidose láctica graves, a quadro moderados, com atraso do desenvolvimento psicomotor/deficiência intelectual e hiperlactacidémia e formas tardias, caracterizadas por ataxia, períodos de fraqueza muscular e/ou intolerância ao exercício físico. No período pré-natal pode já haver manifestações da doença, como malformações do sistema nervoso central (SNC) e/ou restrição do crescimento intra-uterino (RCIU). Um subgrupo de doentes apresenta clínica compatível com síndrome de Leigh, designadamente apneias de causa central.
Os sinais clínicos mais frequentes são o atraso do desenvolvimento psicomotor/deficiência intelectual e a hipotonia, seguidos de epilepsia, microcefalia e ataxia. Esta é mais observada no sexo masculino, podendo ser intermitente, agravada pelo excesso de ingestão de carbohidratos. Outras apresentações menos comuns incluem: disfunção motora com espasticidade, ptose e movimentos coreoatetóides, polipneia por acidémia láctica, especialmente no período neonatal, dismorfismos faciais (mais descritos no sexo feminino), manifestações oculares (atrofia do nervo ótico, nistagmo e/ou estrabismo) e neuropatia periférica.
Exame objectivo
Ao exame objetivo, no recém-nascido podem ser evidentes sinais de RCIU, atraso do desenvolvimento psicomotor, hipotonia ou hipertonia, nistagmo, estrabismo e dismorfismos faciais, como cabeça estreita, bossing frontal, filtro proeminente e ponte nasal larga. Nas formas tardias podem observar-se sinais de disfunção motora, como ataxia, distonia (espasticidade, coreoatetose), ptose ou hemiparesia. A microcefalia pode ser pré ou pos-natal.
Diagnóstico Diferencial
No que refere ao diagnóstico diferencial, para além das outras doenças neurológicas, conta-se as causas mais frequentes de hiperlactacidémia, como sépsis neonatal, choque isquémico ou cardiogénico, cetoacidose diabética, insuficiência hepática e doenças mitocondriais.
Nos doentes com síndrome de Leigh devem ser excluídas doenças da cadeia respiratória mitocondrial, como as causadas por mutações no gene SURF1.
Exames Complementares
Patologia Clínica
O diagnóstico da DPDH passa pelo reconhecimento dos sintomas desta doença, especialmente o envolvimento neurológico precoce, associado a elevação do lactato, do piruvato (com relação lactato/piruvato normal ou diminuída) e da alanina nos fluidos biológicos, a constatação de défice da atividade do complexo da PDH e o estudo genético (2).
Imagiologicamente estão descritas alterações do SNC como a ventriculomegalia (a mais frequente), seguida da hipoagenesia ou agenesia do corpo caloso e a síndrome de Leigh (caraterizada por lesões bilaterais e simétricas dos gânglios da base, tronco e cerebelo). Anomalias dos gânglios da base, tronco cerebral e cerebelo foram igualmente descritas isoladamente em alguns doentes (1).
O estudo enzimático pode ser realizado em fibroblastos ou em outros tecidos, como o músculo, fígado e leucócitos.
O estudo genético permite confirmar o diagnóstico, de forma definitiva.
Não parece haver relação estatisticamente significativa entre o tipo de mutação identificada e a atividade enzimática residual, concentração máxima de lactato ou sobrevivência dos doentes (1).
Tratamento
Nas situações de acidémia lática grave, o tratamento passa pela administração de substâncias alcalinas, como o bicarbonato de sódio.
O tratamento de base da DPDH compreende a estimulação do complexo PDH e/ou o fornecimento de fontes alternativas de energia para o cérebro.
A suplementação com tiamina (vitamina B1), em doses de miligramas a mais de 1 grama por dia, pode ser benéfica em alguns casos, especialmente nos défices da subunidade E1 alfa. Outros cofatores que poderão ser equacionados são a carnitina e o ácido lipóico.
O dicloroacetato, um ativador da PDH pode ser igualmente utilizado quer nas situações agudas de acidose láctica quer cronicamente (oral, em doses iguais ou superiores a 25mg/Kg/dia). No entanto, para além de ser de aquisição difícil, está associado a neurotoxicidade periférica.
A dieta cetogénica é considerada segura e com bons resultados a longo prazo em doentes com DPDH (5). O aumento da ingestão de gorduras em detrimento dos carbohidratos permite que os corpos cetónicos derivados da oxidação de ácidos gordos sejam um fuel cerebral alternativo à glicose. O efeito positivo da dieta verifica-se especialmente em doentes com manifestações iniciais e designadamente na epilepsia, ataxia, distúrbios do sono, desenvolvimento da linguagem, interação social e frequência de hospitalizações. A introdução precoce da dieta em doentes com início de sintomas na primeira infância poderá prevenir lesões metabólicas cerebrais (5).
Dada a raridade da doença, na eventual suspeita de DPDH deve ser feito o contacto/ referenciação com um Centro de Referência de Doenças Hereditárias do Metabolismo, de modo a que o diagnóstico, tratamento e seguimento do doente sejam os mais adequados, de acordo com a norma 012/2017 da Direção Geral da Saúde (Abordagem Diagnóstica e Critérios de Referenciação de Doenças Hereditárias do Metabolismo em Idade Pediátrica e no Adulto, em: dqs@dgs.min-saude.pt).
Evolução
A DPDH tem prognóstico variável, sendo que é desfavorável na maioria dos casos. As formas neonatais graves podem ser fatais na primeira infância. Mais de um terço dos casos descritos com esta doença faleceram na primeira infância, com uma idade média entre os dois e os três anos (2.70+/-4,66 anos) (1).
Os doentes com formas clínicas ligeiras apresentam função cognitiva normal ou borderline e têm prognóstico favorável (3).
A maioria dos casos conhecidos de DPDH são referentes a defeitos na subunidade E1 alfa, associada a hereditariedade ligada ao cromossoma X, estando descritas algumas correlações genótipo/fenótipo associadas ao gene PDHA1. Mutações neste gene com envolvimento do aminoácido na posição 378 são particularmente letais. Pelo contrário, mutações que levam à substituição dos aminoácidos 263 e 302 (a maioria descrita em doentes do sexo masculino) têm melhor prognóstico (1).
A história natural da doença tende a ser similar em ambos os sexos. No entanto, a proporção de falecimentos no sexo masculino é superior, o que poderá refletir o impacto da lionização do alelo PDHA1 (1). De forma inexplicável, os casos de falecimento no sexo feminino tendem a ter início de doença mais precoce e a falecer em idade mais jovem.
Recomendações
O seguimento destes doentes deve ser feito por equipas multidisciplinares, que incluam médicos e nutricionistas com experiência em Doenças Hereditárias do Metabolismo e em Neuropediatria.
Deve ser feita a monitorização do cumprimento da terapêutica, da evolução da doença e o doseamento regular dos níveis plasmáticos de lactato. Nos doentes sob dieta cetogénica, deve ser igualmente realizada uma monitorização regular dos corpos cetónicos e dos possíveis efeitos adversos associados à dieta.
Simultaneamente, deve ser feita a vigilância de eventuais complicações, o que inclui a avaliação oftalmológica regular e a monitorização do desenvolvimento psicomotor/ intelectual e rendimento escolar. Tratando-se de uma doença crónica, com importante impacto na qualidade de vida, os aspetos psico-sociais no seguimento dos doentes e famílias não devem ser descurados.
Deve ser ainda solicitada consulta de Genética com vista ao aconselhamento genético da família. Este visa a avaliação do risco de recorrência da doença e o esclarecimento de eventuais possibilidades reprodutivas numa futura gestação do casal, nomeadamente a de diagnóstico pré-natal (DPN) molecular orientado ou diagnóstico genético pré-implantação (DGPI).
Bibliografia
- Patel KP, O’Brien TW, Subramony SH, Shuster J, Stacpoole PW. The spectrum of pyruvate dehydrogenase complex deficiency: Clinical, biochemical and genetic features in 371 patients. Molecular Genetics and Metabolism. 2012;105:34-43.
- De Meirleir LJ, Garcia-Cazorla A, Brivet M (2016) Disorders of Pyruvate Metabolism and the Tricarboxylic Acid Cycle. In: Saudubray JM, van den Berghe G, Walter JH (eds) Inborn metabolic diseases: diagnosis and treatment. Springer, Heidelberg
- Steller J, Gagus JJ, Gibbs LH, Hasso AN, Kimonis VE. Mild phenotype in a male with Pyruvate Dehydrogenase Complex Deficiency Associated with novel hemizygous in-frame duplications of the E1α subunit gene (PDHA1). Neuropediatrics 2014;45:56-60
- Soreze Y, Boutron A, Habarou F, Barnerias C, Nonnenmacher L, Delpech H, Mamoune A, Chrétien D, Hubert L, Bole-Feysot C, Nitschke P, Correia I, Sardet C, Boddaert N, Hamel Y, Delahodde A, Ottolenghi C, Lonlay P. Mutations in human lipoyltransferase gene LIPT1 cause a Leigh disease with secondary deficiency for pyruvate and alpha-ketoglutarate dehydrogenase. Orphanet Journal of Rare Diseases 2013;8:192
- Sofou K, Dahlin M, Hallbook T, Lindefeldt M, Viggedal G, Darin N. Ketogenic diet in pyruvate dehydrogenase complex deficiency: short and long-term outcomes. 2017 Jan 18;40:237-245
Deseja sugerir alguma alteração para este tema?
Existe algum tema que queira ver na Pedipedia - Enciclopédia Online?