

Icterícia e colestase neonatal
Introdução
Definição
A icterícia é a coloração amarela da pele e escleróticas. Bioquimicamente, corresponde a uma elevação da bilirrubina no sangue.
A colestase é um estado patológico em que há redução da formação de bile ou do seu fluxo. Consequentemente há retenção de substâncias normalmente excretadas na bile (bilirrubina, ácidos biliares, colesterol). Nem todas as substâncias normalmente excretadas na bile são retidas na mesma proporção nas várias doenças colestáticas (exemplo: em algumas doenças pode haver grande retenção de ácidos biliares enquanto a retenção de bilirrubina é ligeira, e vice-versa).
A colestase clínica define-se pela presença de bilirrubina conjugada > 1 mg/dl (1).
Epidemiologia
A icterícia é um problema comum que afeta cerca de 60% dos recém-nascidos (RN), e na maioria dos casos é causada por condições benignas e auto-limitadas (icterícias, fisiológica, do aleitamento, ou do leite materno); a última pode ser responsável por icterícia prolongada (>14 dias), por vezes até aos 2-3 meses de idade. A icterícia prolongada é um problema relativamente comum, afetando até 15% dos RN.
A colestase neonatal é uma entidade rara (1/2500, 0,04%) com grande morbilidade e mortalidade, com muitos doentes sobrevivendo na dependência de transplante hepático.
Fisiopatologia
A imaturidade das vias enzimáticas do fígado e da circulação enterohepática dos ácidos biliares predispõe o RN (particularmente o pretermo) para um estado de “colestase “fisiológica”.
Esta “colestase fisiológica” manifesta-se por níveis séricos mais elevados e por um perfil de ácidos biliares diferente do das crianças e adultos, mas com valores de bilirrubina conjugada
Os valores normais dos biomarcadores de colestase (bilirrubina conjugada e ácidos biliares), ainda não são bem conhecidos no RN, particularmente no pretermo. Também os perfis evolutivos destes biomarcadores de acordo com a idade gestacional ainda não foram completamente descritos.
A GGT, considerada um biomarcador de lesão das vias biliares, actualmente é considerada também um biomarcador de stress oxidativo.
Estudos recentes (2) procurando fatores de risco associados ao desenvolvimento de colestase clínica no RN, tais como prematuridade, baixo peso ao nascimento, asfixia/hipoxia, sépsis, enterocolite necrotizante, cirurgia, e nutrição parentérica, levantam a hipótese da contribuição do stress oxidativo para a patogénese da colestase neonatal.
Etiologia
O diagnóstico de entidades subjacentes é difícil devido ao seu número, raridade e complexidade (Tabela 1). É muito importante também nos casos fatais, de modo a oferecer às famílias um diagnóstico pré-natal para futuras gestações.
Em séries antigas, a hepatite neonatal idiopática (HNI) foi a causa mais comum de colestase neonatal, com uma incidência de 1/4800 a 1/9000 nados-vivos. No entanto, com a descoberta de etiologias específicas que partilham o fenótipo da HNI, e com métodos de diagnóstico mais avançados, a incidência de HNI diminuiu substancialmente. As entidades mais frequentemente identificadas são a atresia das vias biliares (25%-35%), as doenças genéticas (25%), as doenças metabólicas (20%), e a deficiência de alfa-1-antitripsina (10%).
No entanto, enquanto a HNI diminuiu, a “colestase neonatal transitória multifatorial” tornou-se um dos subgrupos importantes, caracterizando-se por início precoce, ausência de causa subjacente conhecida, normalização dos parâmetros clínicos e bioquímicos durante o seguimento, e uma história de alguns fatores de risco neonatais (incidência no RN pretermo de 2,2-4,5%, com fatores de risco em 13,7%) (2). Neste subgrupo a abordagem do diagnóstico das entidades subjacentes é de difícil decisão.
História Clínica
Apresentação Clínica
A apresentação clínica da colestase neonatal pode variar de acordo com a entidade subjacente. No entanto, os achados mais comuns num RN / lactente com colestase são a icterícia, a colúria, e a acolia fecal.
Em relação à icterícia é importante caracterizá-la tendo em conta o momento em que se inicia e como evolui. A icterícia pode iniciar-se nos primeiros dias (ou mesmo nas primeiras 24h) de vida e prolongar-se >14 dias, pode manifestar-se nos primeiros dias de vida seguida de um intervalo livre e depois recorrer mais tardiamente (> 7 dias), ou manifestar-se apenas mais tardiamente (> 7 dias). No primeiro caso a colestase pode instalar-se em qualquer momento da evolução da icterícia, sendo importante ter em conta que a icterícia pode diminuir ao longo das primeiras semanas de vida, à medida que a percentagem de bilirrubina indireta diminui, dando assim uma falsa impressão de que está a resolver. Nos casos em que a icterícia é de instalação mais tardia, depois (ou não) de um intervalo livre, a suspeita da colestase surge mais facilmente.
A colúria é frequente como indicador de hiperbilirrubinemia conjugada, pode ser facilmente confirmada por um exame de urina com tira-teste, mas a negatividade do teste não a exclui.
Por outro lado, a presença de fezes acólicas é sugestiva, mas não diagnóstica, de obstrução biliar extra-hepática, uma vez que esta pode também estar presente na colestase intra-hepática grave. Ao contrário, a presença de fezes pigmentadas sugere a permeabilidade da árvore biliar extra-hepática e, geralmente, torna a atresia biliar improvável. Nas fases iniciais da atresia biliar, as fezes podem ser normais ou pigmentadas de forma intermitente e, por isso, é muito importante que a cor das fezes seja avaliada de forma seriada em todos os RN/lactentes ictéricos. O cartão de cor das fezes é um método validado para este efeito (3).
Na exploração dos dados clínicos é também importante dar relevância à presença ou ausência de outros sinais/sintomas, tais como: distúrbios alimentares (recusa alimentar/vómitos), evolução ponderal, febre/hipotermia, convulsões ou equivalentes, e manifestações hemorrágicas (na pele, no coto umbilical, gastrointestinais ou intracranianas).
É fundamental explorar a história familiar e alguns aspetos dos antecedentes pessoais. A existência ou não de consanguinidade parental ou de irmãos afetados aponta para a suspeita de doenças hereditárias (metabólicas ou outras). É importante também a observação de eventuais estigmas físicos nos pais (síndrome de Alagille). A história obstétrica pode sugerir infeção materna (infeção TORCHS, hepatite B) ou colestase da gravidez (colestase intra-hepática familiar progressiva, PFIC3). Assume particular relevo a exploração de fatores de risco para o desenvolvimento de colestase multifatorial transitória (2).
No exame objetivo é importante dar relevância ao aspeto geral, nutricional (baixo peso ao nascer ou restrição de crescimento intrauterino, má evolução ponderal, desnutrição), sinais vitais (instabilidade hemodinâmica, febre/hipotermia), icterícia, e manifestações hemorrágicas (petéquias, sufusões, sangramento pelo coto umbilical ou locais de punção). Podem estar presentes a microcefalia ou dismorfias (fácies peculiar, ou outras), bem como sopros cardíacos (estenoses pulmonares periféricas ou anomalias cardíacas estruturais). O exame do abdómen pode evidenciar a presença de organomegalias (fígado e baço), ou massas palpáveis (quisto do colédoco), e ascite. Devem ser pesquisadas anomalias oculares (cataratas, corioretinite, macha côr de cereja, embriotoxon posterior, coloboma ocular, hipoplasia do nervo óptico), e neurológicas (irritabilidade/letargia, hipotonia, convulsões ou nistagmo). A observação direta das fezes faz parte do exame objectivo.
Diagnóstico Diferencial
Abordagem diagnóstica
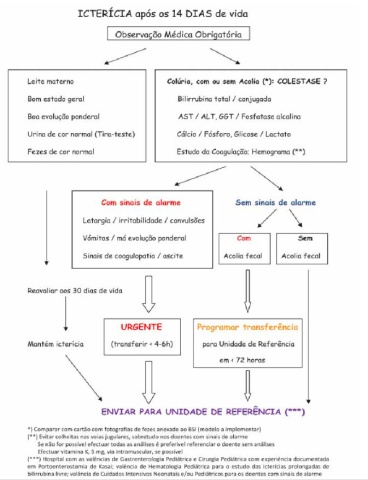
Perante um RN / lactente ictérico a primeira abordagem deve classificar a icterícia como colestática ou de bilirrubina livre. O diagnóstico de colestase deve ser suspeitado imediatamente na presença de colúria, acolia fecal, ou outros sinais/sintomas de doença, e sempre que uma icterícia se prolonga para além dos 14 dias de vida. A confirmação faz-se pelo doseamento sérico das bilirubinas, total e conjugada.
Outros marcadores bioquímicos podem estar aumentados em RN / lactentes com colestase, mas eles não têm valor diagnóstico nem prognóstico: um aumento das transaminases (ALT, AST) indica uma lesão hepatocelular inespecífica; a gamma glutamil-transpeptidase (GGT) é uma enzima do epitélio biliar cujo aumento está associado com doenças colestáticas que envolvem as vias biliares intra ou extrahepáticas (atresia biliar, α 1-antitripsina, síndroma de Alagille, etc), mas também com stress oxidativo (citopatias mitocondriais).
O estudo da coagulação e o doseamento sérico das proteínas totais e albumina são marcadores das funções da síntese hepática. A glicemia é um marcador da manutenção da homeostasia da glicose. O atingimento destas duas funções (síntese e homeostasia da glicose) indicia um maior grau de gravidade da lesão hepática.
O diagnóstico da entidade subjacente é muito complexo, e o risco de investigação inadequada, invasiva e espoliadora é grande. O conhecimento de que um grande número de fatores de risco, sobretudo no RN pretermo, pode ser responsável por si só pela colestase, recomenda enorme ponderação nestes casos. A abordagem deve apoiar-se na exploração minuciosa dos dados da anamnese e do exame objetivo, e na realização de exames complementares orientada pela clínica.
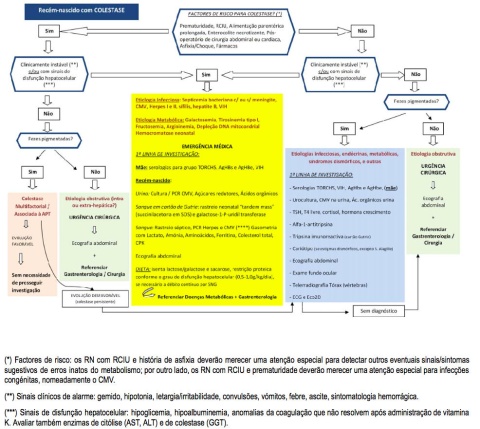
A abordagem que nos parece mais eficiente está resumida nos algoritmos de decisão (algoritmos 1 e 2). De notar que para a colestase associada a alimentação parentérica não existe nenhum marcador específico, pelo que este diagnóstico será sempre de exclusão. Nos casos restantes, a estratégia seguida é a de em primeiro lugar identificar os RN que apresentem sinais de alarme (fezes despigmentadas ou acolia fecal), e RN clinicamente instáveis (sinais de sepsis: letargia, irritabilidade, gemido, febre, vómitos) e/ou com sinais de insuficiência hepatocelular (ascite, coagulopatia).
- Os primeiros, embora não corram risco de vida no imediato, devem ser considerados “urgências cirúrgicas”, pois o atraso no reconhecimento, sobretudo da atresia das vias biliares, pode comprometer de forma irremediável o seu prognóstico a longo prazo. Nestas situações não se deve perder tempo com a realização de exames de 2ª linha, e deve ser efetuado um contacto para transferência para centro de referência, em tempo útil.
- Os segundos devem ser considerados “emergências médicas”, pois correm risco de vida no imediato, e podem falecer enquanto se procede á investigação etiológica. Neste grupo iremos encontrar sobretudo doenças infeciosas (sépsis bacteriana, ITU a E.coli, infeção a Herpes, sífilis, …) e/ou erros inatos do metabolismo (galactosemia, tirosinemia, fructosemia, CDG tipo 1b).
Na primeira abordagem deve ser dada prioridade às doenças curáveis ou tratáveis. Sempre que possível, na suspeita de infeção congénita ou perinatal, os exames podem ser efetuados na mãe, poupando o RN a uma maior espoliação (ex: sífilis, VIH, VHB).
Após esta primeira linha de investigação consideramos que deve ser solicitada a intervenção de especialistas em Gastrenterologia/Hepatologia pediátrica e/ou Doenças Metabólicas, para orientação personalizada. Esta recomendação tem por objetivos evitar a invasividade e a espoliação excessiva de RN em estado crítico, dado que isso conduzirá à necessidade de transfusão de glóbulos vermelhos, com a consequente sobrecarga em ferro (mais uma agressão ao fígado doente) reduzir custos, e sobretudo aumentar a eficácia diagnóstica.
Nos RN com colestase sem sinais de alarme propõe-se uma 1ª linha de investigação dirigida às patologias mais comuns, e os exames complementares menos invasivos e dispendiosos. A investigação subsequente deverá ser programada caso a caso, por especialistas na área. Neste grupo, poderão estar incluídas as doenças dos peroxissomas (Zellweger e Refsum) cujo diagnóstico é suspeitado também pelas dismorfias e/ou anomalias do exame neurológico, entre outras. Nos casos de “colestase de carácter transitório”, poderá ser necessário rastrear algumas doenças metabólicas com expressão mais tardia em outros órgãos e sistemas (ex: Doença de Niemann-Pick tipo C, Citrulinemia tipo II). De notar que para efetuar o diagnóstico de erros inatos do metabolismo sem tratamento específico, mas com possibilidade de diagnóstico pré-natal, também podemos recorrer a exames programados para o “pós-mortem” imediato.
Exames Complementares
Os exames complementares de 1ª linha englobam as bilirrubinas total e conjugada, a GGT, marcadores de lesão hepatocelular (AST/ALT), marcadores da síntese hepática (coagulação, proteínas totais e albumina) e homeostasia da glicose (glicemia) e do metabolismo fosfocálcico (fosfatase alcalina, cálcio e fósforo). Incluem ainda parâmetros de infeção (hemograma e proteína C reativa), bem como alguns exames pouco (ou nada) invasivos, de fácil acesso, e que podem orientar para uma entidade subjacente entre as mais comuns: pesquisa de substâncias redutoras urinárias, urocultura, doseamento de alfa-1-antitripsina, serologias do grupo TORCHS e hepatite B (na mãe), e ecografia abdominal.
A ecografia abdominal é uma ferramenta importante na abordagem diagnóstica inicial e é o estudo de imagem mais útil. Ela pode avaliar o tamanho e a aparência do fígado e vesícula biliar - incluindo a visualização de cálculos biliares e lama biliar. Pode estabelecer o diagnóstico de quisto do colédoco ou demonstrar uma vesícula biliar pequena ou ausente, o que pode sugerir (sem confirmar ou excluir) a atresia biliar. A descoberta do sinal do cordão triangular, uma área ecogénica da “porta hepatis” devido a um cone de tecido fibroso, é específico para atresia biliar, mas a sua visualização exige um radiologista experiente. A dilatação do ducto biliar comum não é vista na atresia biliar e sugere uma obstrução distal ou uma forma fruste de quisto do colédoco.
A cintilografia hepatobiliar com tecnécio-99 marcado com ácido imunodiacético (IDA) caiu em desuso pois quando as fezes acólicas são vistas por um observador experiente este exame acrescenta pouca informação.
Em relação à colangio-ressonância magnética os dados disponíveis são ainda insuficientes para a recomendar rotineiramente.
A biópsia hepática pode ser um auxiliar de diagnóstico no caso de certas doenças específicas, tais como o défice de α 1-antitripsina ou a colangite esclerosante neonatal, mas só deve ser efetuado depois de outros exames terem possibilitado uma primeira orientação diagnóstica.
Tratamento
Algumas das entidades subjacentes têm tratamento específico, médico ou cirúrgico. O quisto do colédoco e a atresia das vias biliares têm tratamento cirúrgico, e sobretudo nesta última o prognóstico depende da precocidade com que a cirurgia é executada, e das capacidades e da experiência da equipa. O prognóstico de alguns erros inatos do metabolismo também depende da precocidade do diagnóstico e do tratamento médico específico (dietas especiais, 2-(2-nitro-4- trifluorometilbenzoil) -1,3-ciclohexanediona - NTBC e outros fármacos), particularmente aqueles que se apresentam com sinais de insuficiência hepatocelular. A sobrevida global destes doentes depende também da disponibilidade do transplante hepático para aqueles com evolução para doença hepática terminal.
Mesmo os doentes com patologias sem tratamento específico podem beneficiar de tratamento de suporte, e sendo este fundamental também como ponte para o transplante hepático. Em ambos os casos a eficácia é tanto maior quanto mais cedo o tratamento for instituído.
Os doentes com colestase frequentemente têm esteatorreia e aumento dos gastos energéticos. Portanto, o aporte calórico deve ser aproximadamente 125% da ingestão diária recomendada baseada no peso corporal ideal. Os triglicerídeos de cadeia média (MCT) são mais facilmente absorvidos do que os ácidos gordos de cadeia longa e são a melhor fonte de calorias lipídicas. Na verdade, os MCT são relativamente solúveis em água, não necessitam de solubilização de micelas dos ácidos biliares e podem ser diretamente absorvidos pela circulação portal.
Em RN / lactentes pretermo a nutrição parentérica deve ser descontinuada o mais rapidamente possível e a alimentação entérica (incluindo alimentação trófica) deve ser promovida pois aumenta o fluxo biliar, a contração da vesícula, e a motilidade intestinal. Alguns estudos evidenciam que uma nova emulsão lipídica (SMOFlipid®) contendo uma mistura de óleo de soja, MCT, azeite e óleo de peixe com redução de ácidos gordos omega-6, aumento de ácidos gordos omega-3, e enriquecida em vitamina E, se associa a uma diminuição da GGT sérica, do stress oxidativo, e da retinopatia da prematuridade, em comparação com as emulsões à base de óleo de soja.
A absorção intestinal de vitaminas lipossolúveis (A, D, E e K) requer a presença de ácidos biliares, pelo que são recomendadas doses de pelo menos duas a quatro vezes a dose diária habitual (4). O primeiro défice a instalar-se é o da vitamina K, que surge em alguns dias, pelo que a suplementação deve iniciar-se imediatamente após o diagnóstico da colestase. A suplementação nas outras vitaminas deve continuar pelo menos três meses após a resolução da icterícia, dado haver um atraso até que o fluxo biliar normal seja restabelecido.
O ácido ursodesoxicólico (AUDC), tem sido associado a efeitos benéficos na colestase, e é geralmente usado como terapia de primeira linha para o prurido, colestase induzida pela nutrição parentérica, pós-operatório da atresia biliar, e defice de alfa-1-antitripsina. O seu modo de ação não está completamente compreendido, mas parece ter dois componentes: (a) a substituição do “pool” de ácidos biliares por outros hidrofílicos menos hepatotóxicos, e (b) estimulação do fluxo biliar. A dosagem é de 15 a 20 mg/kg/dia em duas a três doses/dia. O único efeito colateral frequente é a diarreia, que geralmente responde à redução da dose. O AUDC pode ser interrompido quando as bilirrubinas e a GGT estiverem normais.
A rifampicina inibe a absorção de ácidos biliares pelos hepatócitos e induz enzimas microssomais hepáticas. A dosagem recomendada é de 10 mg/kg/dia. Está indicada no tratamento do prurido refratário, mas a função do fígado deve ser monitorizada devido ao seu potencial de hepatotoxicidade.
A fototerapia não está contra-indicada nos RN com colestase, e a bilirrubina conjugada não deve ser subtraída à bilirrubina total na tomada de decisões sobre a realização de exanguíneo-transfusões.
Evolução
A colestase neonatal é globalmente uma entidade nosológica com grande morbilidade e mortalidade. Individualmente o prognóstico depende da entidade subjacente, da precocidade de diagnóstico e da instituição de terapêutica específica (quando ela está disponível), bem como da terapêutica de suporte da colestase; depende também da terapêutica de eventuais co-morbilidades, e da acessibilidade ao transplante hepático.
O reconhecimento precoce da colestase neonatal é essencial para assegurar um tratamento atempado e um melhor prognóstico. Mesmo quando não está disponível um tratamento específico, os doentes beneficiam de tratamento de suporte e da otimização do seu estado nutricional. O desenvolvimento de ferramentas clínicas para melhorar o prognóstico/resultado da colestase neonatal é atualmente um importante tópico de investigação.
Apesar destes progressos, o atraso no diagnóstico permanece um problema em todos os países e algumas estratégias para resolver este problema foram tentadas. No entanto, nada mais que soluções parciais têm sido propostas.
Existem alguns obstáculos ao reconhecimento precoce da colestase neonatal. A principal manifestação, a icterícia, é um problema comum que afeta cerca de 60% dos recém-nascidos, e na maioria dos casos é causada por condições benignas e autolimitadas. Talvez por essa razão, os profissionais de saúde tendam a subestimar o sintoma, especialmente nos casos em que a icterícia não é acompanhada de outros sinais ou sintomas da doença, retardando o diagnóstico.
Por outro lado, muitos profissionais de saúde (médicos e enfermeiros), parecem ter uma grande impreparação para reconhecer a presença de outros sinais de colestase (colúria e acolia fecal) (5).
De acordo com as recomendações atuais, qualquer RN que permaneça ictérico para além dos 14 dias de vida deve ser observado por um médico, e qualquer RN, independentemente da idade, que, além de icterícia apresente outros sinais ou sintomas de doença, incluindo fezes despigmentadas, deve ser imediatamente encaminhado para um centro especializado para uma investigação mais aprofundada.
O rastreio universal da colestase neonatal seria justificado pela incidência de 1/2500 nascidos vivos, e pelo conhecimento de várias entidades subjacentes com tratamento eficaz, cujo prognóstico depende da precocidade com que é iniciado. No entanto, mesmo que fosse encontrado um método fiável, não-invasivo e de baixo custo, o grande intervalo de tempo para a instalação de sinais e sintomas, de acordo com as várias patologias subjacentes, impede que possa ser encontrado um tempo ideal para o aplicar.
O rastreio é viável e tem sido realizado com sucesso, para uma das entidades subjacentes, a atresia das vias biliares, e em países onde a incidência desta o torna custo-efetivo, como acontece nos países asiáticos. Pelo contrário, nos países europeus, onde a atresia das vias biliares é uma doença muito rara (1/18000), a triagem pelo método do cartão de cor das fezes ainda não foi reconhecida como sendo custo-efetiva, apesar das tentativas de alguns autores para o demonstrar.
O rastreio universal de alguns erros inatos do metabolismo está incluído no Programa Português de Rastreio Neonatal.
Recomendações
- Investigar imediatamente todos os RN ictéricos com sinais de sépsis ou de insuficiência hepatocelular (ascite, hemorragias), colúria e/ou fezes despigmentadas, organomegalias, e estigmas dismórficos.
- Investigar todos os RN com icterícia prolongada para além dos 14 dias de vida.
- Enviar para Centro de Referência as situações urgentes, quando os exames de 1ª linha não esclarecem o diagnóstico, e quando é diagnosticada patologia que exija abordagem especializada.
- Não esquecer a suplementação imediata com vitamina K via endovenosa (4).
RN / lactentes internados em Unidades de Cuidados Intensivos Neonatais e Pediátricas
- A colestase de etiologia multifatorial e a associada à alimentação parentérica são as entidades mais frequentes, pelo que se recomenda que a investigação de causas subjacentes seja reduzida ao estritamente necessário, e seja diferido o esclarecimento das patologias não urgentes.
- A realização dos exames de 1ª linha, e de outros exames deverá ser ponderada caso a caso com a ajuda do especialista em Gastroenterologia Pediátrica e/ou Doenças Metabólicas.
- Estes RN/lactentes devem receber uma ingestão calórica cerca de 125% da ingestão dietética recomendada para os saudáveis, com uma preferência por MCT como fonte de lípideos. Na nutrição parentérica de RN pretermos deve ser usada uma emulsão lipídica baseada em óleo de peixe em vez de ser à base de soja.
- Deve ser garantida uma suplementação adequada de vitaminas (4).
- O AUDC deve ser administrado na dose de 20-30 mg/kg/dia, dividida em 2-3 tomas/dia, e descontinuado logo que a colestase esteja resolvida.
- A eritromicina via oral pode ser usada em RN pretermos para estimular a tolerância à nutrição entérica, devido à sua ação procinética.
- A colestase não contra-indica a fototerapia quando valor de bilirrubina não conjugada a recomenda. A bilirrubina conjugada não deve ser subtraída à bilirrubina total na tomada de decisões sobre a realização de exsanguíneo-transfusão.
Glossário
Icterícia fisiológica do RN: aparece após as primeiras 24h e tem uma duração máxima de 7 dias no RN de termo, e de 14 dias no pretermo.
Icterícia patológica do RN: pode apresentar-se nas 1ªs 24h de vida, ser de grande intensidade obrigando a terapêuticas intensivas/invasivas (fototerapia dupla ou exanguíneo-transfusão) ou ser prolongada (> 14 dias).
A icterícia pode classificar-se de acordo com a percentagem da fração de bilirrubina conjugada:
Colestase clínica: hiperbilirrubinemia com fracção conjugada >1 mg/dl. A colestase clínica é sempre patológica, e é uma forma frequente de expressão de muitas doenças subjacentes.
Icterícia de bilirrubina livre: hiperbilirrubinemia com predomínio da fração não conjugada (fração conjugada
Bibliografia
- Fawaz R, Baumann U, Ekong U, Fischler B, Hadzic N, Mack CL, et al. Guideline for the Evaluation of Cholestatic Jaundice in Infants: Joint Recommendations of the North American Society for Pediatric Gastroenterology, Hepatology, and Nutrition and the European Society for Pediatric Gastroenterology, Hepatology, and Nutrition. J Pediatr Gastroenterol Nutr. 2017;64(1):154-68.
- Champion V, Carbajal R, Lozar J, Girard I, Mitanchez D. Risk factors for developing transient neonatal cholestasis. J Pediatr Gastroenterol Nutr. 2012;55(5):592-8.
- Hoshino E, Hayashi K, Suzuki M, Obatake M, Urayama KY, Nakano S, et al. An iPhone application using a novel stool color detection algorithm for biliary atresia screening. Pediatr Surg Int. 2017.
- Dani C, Pratesi S, Raimondi F, Romagnoli C, Task Force for Hyperbilirubinemia of the Italian Society of N. Italian guidelines for the management and treatment of neonatal cholestasis. Ital J Pediatr. 2015;41:69.
- Santos Silva E, Moreira Silva H, Azevedo Lijnzaat L, Melo C, Costa E, Martins E, et al. Clinical practices among healthcare professionals concerning neonatal jaundice and pale stools. Eur J Pediatr. 2017;176(3):361-9.
Deseja sugerir alguma alteração para este tema?
Existe algum tema que queira ver na Pedipedia - Enciclopédia Online?