

Deficiência da ornitina transcarbamílase (OTC)
- Dor de cabeça (cefaleia)
- Alterações de comportamento alimentar
- Alterações do sono e insónias
- Comportamento agressivo
- Delírio
- Demência
- Depressão
- Hiperactividade
- Perda de apetite (Anorexia)
- Problemas de atenção/ concentração
- Atraso de crescimento
- Atraso de desenvolvimento
- Confusão/ desorientação
- Convulsões
- Desmaio/ perda de consciência
- Desnutrição/ caquexia
- Perda de apetite (Anorexia)
- Perda de peso/ Emagrecimento
- Cansaço/ fadiga (astenia)
- Enjôos e náuseas
- Vómitos
- Irritabilidade
- Vómitos
Introdução
Definição
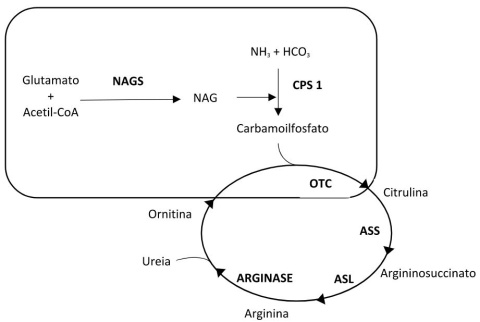
A deficiência da ornitina transcarbamílase (OTC) é um distúrbio mitocondrial do ciclo da ureia, afectando a reacção que catalisa a síntese da citrulina a partir do carbamoilfosfato e da ornitina (1).
O ciclo da ureia é a principal via de eliminação da amónia do organismo e qualquer interferência, especialmente se numa posição inicial do ciclo, como a deficiência de OTC, vai condicionar hiperamoniémia (1).
Epidemiologia
É a doença mais comum do ciclo da ureia, com incidência variando de 1:14.000 a 1:77.000.2 A sua transmissão é ligada ao cromossoma X (gene OTC no Xp11) e são conhecidas cerca de 500 mutações >(1).
História Clínica
Na deficiência de OTC, as manifestações clínicas decorrem maioritariamente da elevação da amónia e sua neurotoxicidade.
Nos homens (hemizigotos), a apresentação é geralmente mais precoce e grave, enquanto as mulheres (heterozigotas) podem ser assintomáticas, desenvolver sintomas tardios ou também ter manifestações graves e precoces (2).
A apresentação no período neonatal é geralmente aguda e rapidamente evolutiva. Tipicamente, um recém-nascido do sexo masculino (raramente do sexo feminino), aparentemente saudável, inicia, ao longo dos primeiros dias de vida, sucção débil, hipotonia, letargia, convulsões, com possível progressão para coma e morte. É frequentemente confundida com sépsis neonatal (3).
Na infância, a deficiência de OTC pode manifestar-se de forma aguda, com episódios de irritabilidade, encefalopatia, ataxia, convulsões, ou de modo discreto e paulatino, através de má evolução estaturo-ponderal, atraso de desenvolvimento psicomotor ou vómitos recorrentes (3).
As crianças mais velhas e os adultos podem exibir episódios recorrentes de encefalopatia, que mimetizam encefalites e intoxicações, ou, de modo mais silencioso, com dificuldades de aprendizagem, sintomas psiquiátricos, enxaqueca, letargia (3).
Nos diferentes grupos etários, é comum haver atingimento hepático, que acaba, por vezes, por ser o ponto motivador da pesquisa diagnóstica. As alterações encontradas podem variar de hepatomegalia, citólise ligeira a insuficiência hepática grave.
Existem dados na anamnese que podem apoiar a suspeita diagnóstica, nomeadamente a presença de: história familiar de mortes de recém-nascidos do sexo masculino com suspeita de sépsis ou causas indeterminadas; antecedentes na família de mulheres com sintomatologia episódica sem etiologia determinada ou coma no pós-parto; história de aversão a alimentos ricos em proteína.
Nas apresentações agudas e recorrentes, é comum os sintomas surgirem no decurso de factores precipitantes, como infecções, vómitos, jejum, sobrecarga proteica, cirurgia, parto, fármacos, os quais podem potenciar uma crise hiperamoniémica, em qualquer idade e independentemente da gravidade da deficiência enzimática (2,4).
Diagnóstico Diferencial
Nos casos de apresentação aguda, o diagnóstico diferencial coloca-se com outras patologias mais frequentes que podem cursar com encefalopatia, como sépsis (principalmente no período neonatal), encefalite, intoxicação por agente externo, acidente vascular cerebral, tumor cerebral, etc.. É, assim, fundamental que perante uma situação inexplicada de alteração de consciência, a par da investigação dessas entidades mais comuns, seja também pensada a deficiência de OTC e requisitado o doseamento de amónia.4
Perante a evidência de hiperamoniémia devem ser considerados outros diagnósticos diferenciais, dos quais se destacam:
- Outros distúrbios do ciclo da ureia: A deficiência de OTC é clinicamente indistinguível dos restantes distúrbios do ciclo da ureia, principalmente dos outros defeitos intramitocondriais, como a deficiência de NAGS e a deficiência de CPS1. Relativamente aos defeitos citosólicos (deficiência de ASS, ASL e arginase), estes cursam geralmente com valores de amónia mais baixos e podem ter associadas outras manifestações características, como a tetraparésia espástica de apresentação precoce (deficiência de arginase) e o cabelo quebradiço (trichorrexis nodosa na deficiência de ASL) (1)
- Acidúrias orgânicas: O nível de hiperamoniémia é menos elevado (geralmente inferior a 200 µmol/L) e predomina a acidose metabólica grave, com cetose acentuada, por vezes, acompanhada de instabilidade glicémica e leucopenia/pancitopenia (4). Nas acidúrias é ainda comum o doente apresentar um cheiro característico, não evidente na deficiência de OTC.
- Defeitos da β-oxidação dos ácidos gordos: É característico a hipoglicemia hipocetótica, eventualmente acompanhada de acidose e hiperamoniémia ligeira a moderada. Pode ocorrer atingimento cardíaco (cardiomiopatia e arritmias), muscular (rabdomiólise) e hepático (4)
- Hiperamoniémia transitória: Pode surgir nos recém-nascidos prematuros, provavelmente devido à patência do ducto venoso que possibilita shunt do sangue portal. Os valores de glutamina encontram-se dentro dos parâmetros da normalidade (3)
- Doença grave no período neonatal (4)
- Insuficiência hepática
- Fármacos: Valproato de sódio, corticoesteróides (4)
- Falsos positivos: Colheita e transporte inadequados e processamento tardio da amostra (3)
Exames Complementares
Amónia: Está elevada, principalmente nas descompensações, podendo atingir valores> 500-1000 µmol/L. (1)
Equilíbrio ácido-base: No início existe alcalose respiratória, contudo, a avaliação é, muitas vezes, feita numa fase em que já está instalada acidose metabólica. (3)
Marcadores de citólise e função hepática
Cromatografia de aminoácidos no plasma: A glutamina e alanina estão geralmente aumentadas e a citrulina e a arginina diminuídas. (4)
Ácido orótico na urina: O carbamoilfosfato acumulado vai derivar para a via das pirimidinas e levar à produção excessiva de ácido orótico. (1)
Estudo enzimático: Por ser necessária biópsia hepática ou de intestino e ser inconclusivo na mulher, é um exame cada menos requisitado para confirmação diagnóstica.
Estudo molecular do gene OTC: É o exame de escolha para confirmação diagnóstica. Por sequenciação são identificados 80% dos casos; nos restantes ponderar MPLA, array-CGH. (1)
Prova do alopurinol: Pode ser útil na impossibilidade de identificar a alteração genética. (1)

Tratamento
Urgência
Perante um doente com diagnóstico ou suspeita de deficiência de OTC, o tratamento de uma descompensação aguda deve ser iniciado de imediato, uma vez que o prognóstico está intimamente relacionado com o controlo da hiperamoniémia. (2) Deve ser consultada informação de medidas de emergência no Cartão de Pessoa com Doença Rara e/ou estabelecido contacto com Centro de Referência de Doenças Hereditárias do Metabolismo. (5) No entretanto, podem ser iniciadas algumas medidas terapêuticas: (1-4)
- Suspender o aporte de proteínas, não mais do que 24 a 48h, de modo a prevenir o catabolismo das proteínas endógenas. Reiniciar quando amónia
- Promover o anabolismo, fornecendo cargas calóricas elevadas (120% das necessidades energéticas ajustadas à idade):
- Perfusão de glicose a 10% por via periférica (por via central podem ser oferecidas doses mais elevadas). No recém-nascido, manter cargas de 10 a 15mg/kg/min de glicose. Associar cloreto de sódio e potássio à perfusão.
- Se hiperglicemia, não reduzir o aporte de glicose mas associar perfusão de insulina.
- Administrar lipídeos por via endovenosa (1 a 3 g/kg/d), se necessário reforço calórico.
- Prevenir o vómito com medicação antiemética.
- Fármacos:
Nas descompensações agudas, são utilizados fármacos depuradores de amónia (benzoato de sódio e/ou fenilbutirato de sódio) e arginina.- Benzoato de sódio: Depuração de amónia através da conjugação com a glicina. Pode ser administrada por via oral ou endovenosa.
Bólus de 250 mg/kg (se >20kg – 5,5 g/m2) nas primeiras 2h; manutenção: 250 - 500 mg/kg/d (se >20Kg – 5,5 g/m2/dia) nas 24h seguintes (máximo de 12 g/dia). - Fenilbutirato de sódio: Depuração de amónia através da conjugação com a glutamina. Administração por via oral.
Bólus de 250 mg/kg (se> 20kg – 5,5 g/m2) nas primeiras 2h; manutenção: 250 - 500 mg/kg/d (se >20Kg – 5,5 g/m2/dia) nas 24h seguintes (máximo de 12 g/dia). - Mistura de fenilacetato de sódio e benzoato de sódio: Recomendada quando forem necessários os dois fármacos depuradores de amónia e só for possível administração por via endovenosa.
Bólus de 2,5 mL/kg (se> 20 kg – 55 ml/m2) nas primeiras 2h; manutenção: 2,5ml/kg/d (se > 20Kg – 55 ml/m2) nas 24h seguintes. - Arginina: Pode ser administrada por via oral ou endovenosa.
Bólus de 250 - 400mg/kg nas primeiras 2h; manutenção: 250 mg/kg/d (máximo de 12 g/dia).
- Benzoato de sódio: Depuração de amónia através da conjugação com a glicina. Pode ser administrada por via oral ou endovenosa.
- Técnicas de depuração extra-renal: A instituir se amónia> 500 µmol/L ou ausência de resposta com as medidas instituídas. Os métodos mais eficazes são a hemodiálise e a hemodiafiltração.
- Doseamento de amónia cada 3h.
- Transferência para um centro com experiência no tratamento de doenças do ciclo da ureia.
Manutenção
O tratamento de manutenção tem de ser individualizado caso a caso, de modo a ser garantido o crescimento e o desenvolvimento adequados e minimizado o número de descompensações.
O tratamento conservador geralmente implica: (1-3)
- Dieta com aporte proteico controlado, respeitando os mínimos de segurança recomendados pela FAO/OMS, e, de forma a permitir o adequado crescimento de desenvolvimento psicomotor, é, muitas vezes, necessário fornecer suplementos de aminoácidos essenciais, vitaminas e oligoelementos.
- Fármacos depuradores de amónia:
- Benzoato de sódio: 200 a 250 mg/kg/dia, divididos em 3 doses.
- Fenilbutirato de sódio: 200 a 250 mg/kg/dia, divididos em 3 doses.
- Fenilbutirato de glicerol: 5 to 12.4 g/m²/dia.
- Suplementos de arginina ou citrulina: 100 a 200 mg/kg/dia.
No caso de doentes com deficiência de OTC grave, com crises de hiperamoniémia recorrentes, deve ser ponderada a orientação atempada para transplante hepático, antes que estejam instaladas alterações neurológicas. (1)
Evolução
A hiperamoniémia pode ser fatal ou produzir sequelas neurológicas, como atraso mental, dificuldades de aprendizagem e distúrbios psiquiátricos. Os casos de apresentação grave, com início no período neonatal, têm maior risco de mortalidade e de lesões, em consequência não só da magnitude da hiperamoniémia, mas sobretudo do tempo de exposição a esses níveis e da instalação do aumento de pressão intracraniana. Deste modo, o prognóstico está dependente da capacidade de prevenir a ocorrência de crises de hiperamoniémia e do controlo da sua gravidade e duração. (1)
É fundamental a manutenção de vigilância clínica e analítica para avaliação de evolução, nomeadamente de eventuais défices nutricionais decorrentes da dieta controlada, os quais podem condicionar atraso de crescimento, obesidade, atraso pubertário, alterações esqueléticas e de tegumento. Estes doentes/famílias necessitam de apoio de equipa multidisciplinar alargada que os oriente nos diferentes níveis individual, familiar, social. (5)
Glossário
Hiperamoniémia: valor de amónia >50 μmol/L (>90μg/dl) nas crianças e nos adultos e >100 μmol/L (>180μg/dl) nos recém-nascidos.2
NAG: N-acetilglutamato
NAGS: N-acetilglutamato sintetase
CPS1: Carbamoil fosfato sintetase 1
ASS: Argininossuccinato sintetase
ASL: Argininossuccinato liase
MLPA: multiplex ligation dependente probe amplification
array-CGH: array comparative genomic hybridization
Bibliografia
- Saudubray JM, Baumgartner M, Walter J. Inborn Metabolic Diseases – Diagnosis and Treatment, 6th Edition: Springer; 2016.
- Secção de Doenças Hereditárias do Metabolismo da Sociedade Portuguesa de Pediatria. Hiperamoniémia em idade pediátrica: Protocolo de avaliação diagnóstica e orientação terapêutica iniciais. 2016. Disponível em: http://www.spp.pt/conteudos/default.asp?ID=528
- Lichter-Konecki, U, Caldovic L, Morizono H, Simpson K. Ornithine Transcarbamylase Deficiency; [Last Update: April 14, 2016]. GeneReviews® [Internet]. Disponível em: https://www.ncbi.nlm.nih.gov/books/NBK154378/
- Häberle J, Boddaert N, Burlina A, Chakrapani A, Dixon M, Huemer M et al. Suggested guidelines for the diagnosis and management of urea cycle disorders. Orphanet J Rare Dis. 2012; 7:32.
- Direção-Geral da Saúde. Norma de Abordagem Diagnóstica e Critérios de Referenciação de Doenças Hereditárias do Metabolismo em Idade Pediátrica e no Adulto. Data de publicação: 12/07/2017. Disponível em: https://www.dgs.pt/directrizes-da-dgs/normas-e-circulares-normativas.aspx?cachecontrol=1502105754031
Deseja sugerir alguma alteração para este tema?
Existe algum tema que queira ver na Pedipedia - Enciclopédia Online?